The CXCR4 project is a multifaceted program focusing on developing small molecules for therapeutic intervention in a number of diseases including cancer, stem cell transplant, neutropenia and HIV infection. The biological roles of this G-protein coupled receptor (GPCR) are both far reaching and significant. The main process that this receptor regulates is one on a cellular level termed chemotaxis, which is the movement of cells within the human body. These include regulation of immune cells, propagation of cancer cells, and facilitating HIV entry. Furthermore, since this receptor involves controlling cell location, it can be coupled together with other therapies to enhance therapeutic effects. THE LRG has developed CXCR4 antagonists that can be administered orally and are therapeutically effective in a number of mouse models representing various disease states.
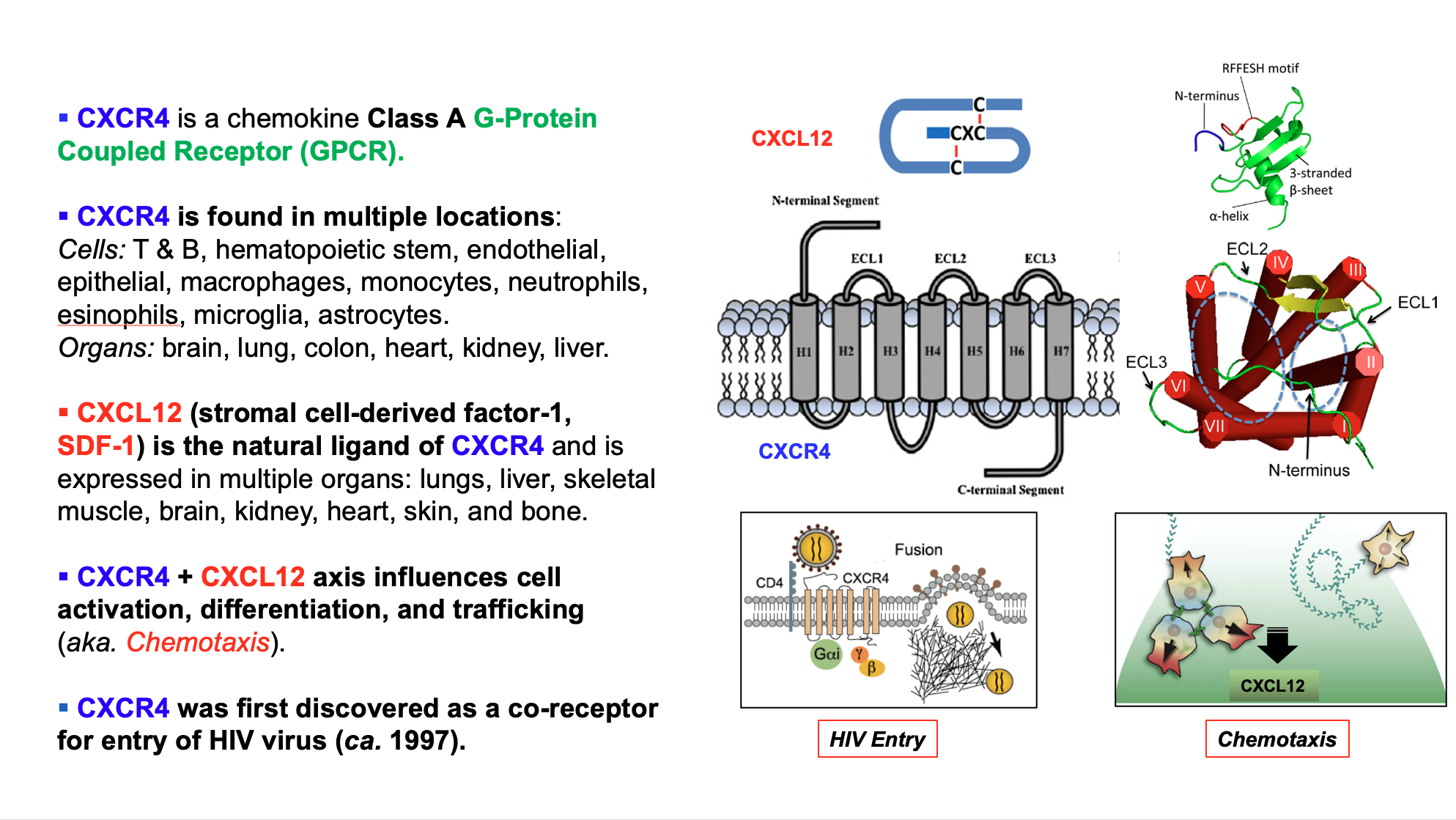
CXCR4 (CXC chemokine receptor 4) is a seven transmembrane G protein-coupled chemokine receptor. Its only endogenous ligand is CXCL12 (CXC chemokine ligand 12, also known as stromal cell-derived factor 1 (SDF-1)). CXCL12 regulates the trafficking of CXCR4+ leukocytes and hematopoetic stem cells under normal physiological conditions (homeostasis). CXCR4 is expressed in T cells, neutrophils, mast cells, eosinophils, basophils, B cells, monocytes/macrophages, dendritic cells, endothelial cells. CXCR4 also plays a crucial role in pathological conditions, such as cancer, HIV/AIDS, and autoimmune diseases.
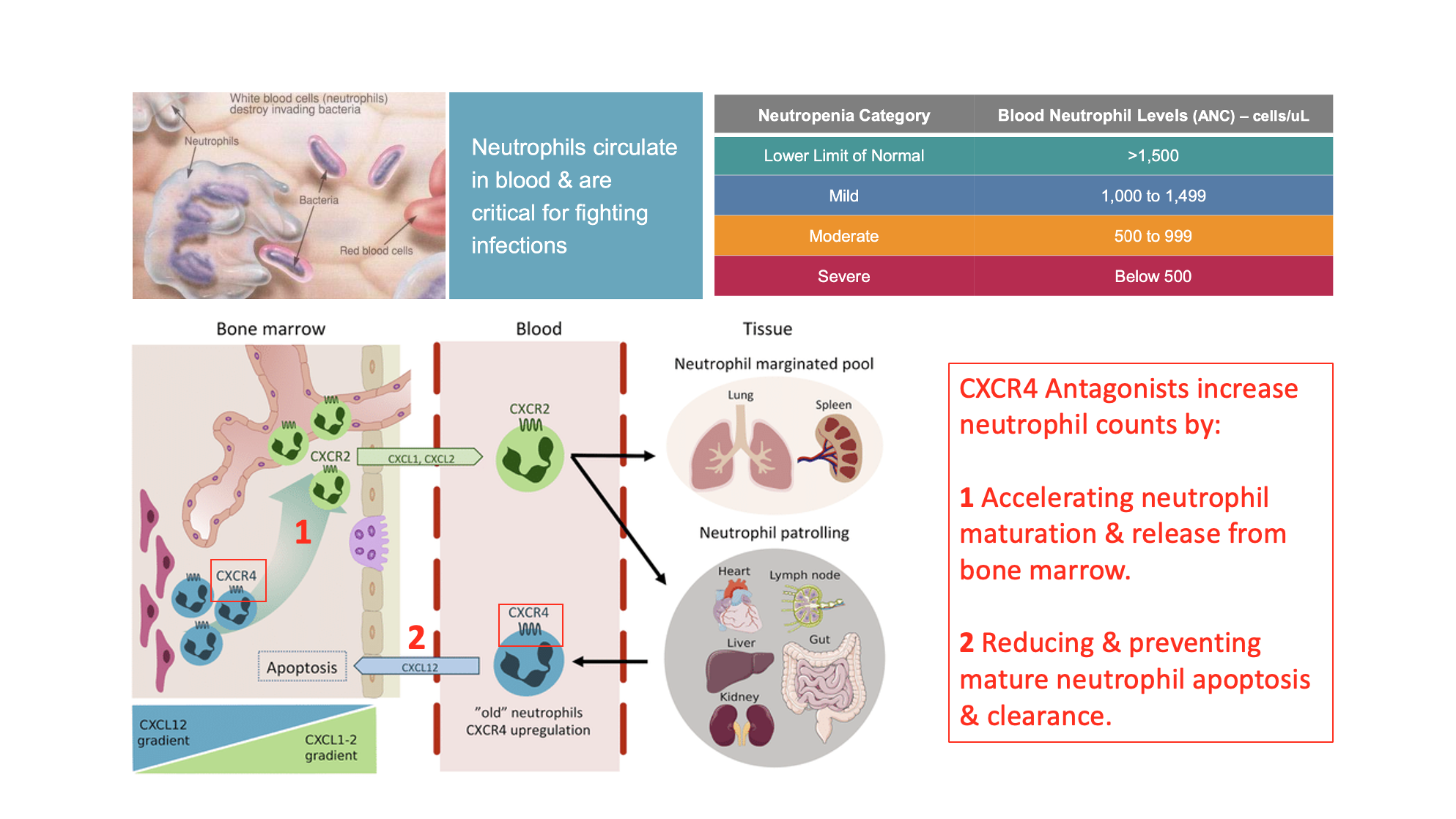
The CXCR4 biology is summarized in the following schematic:
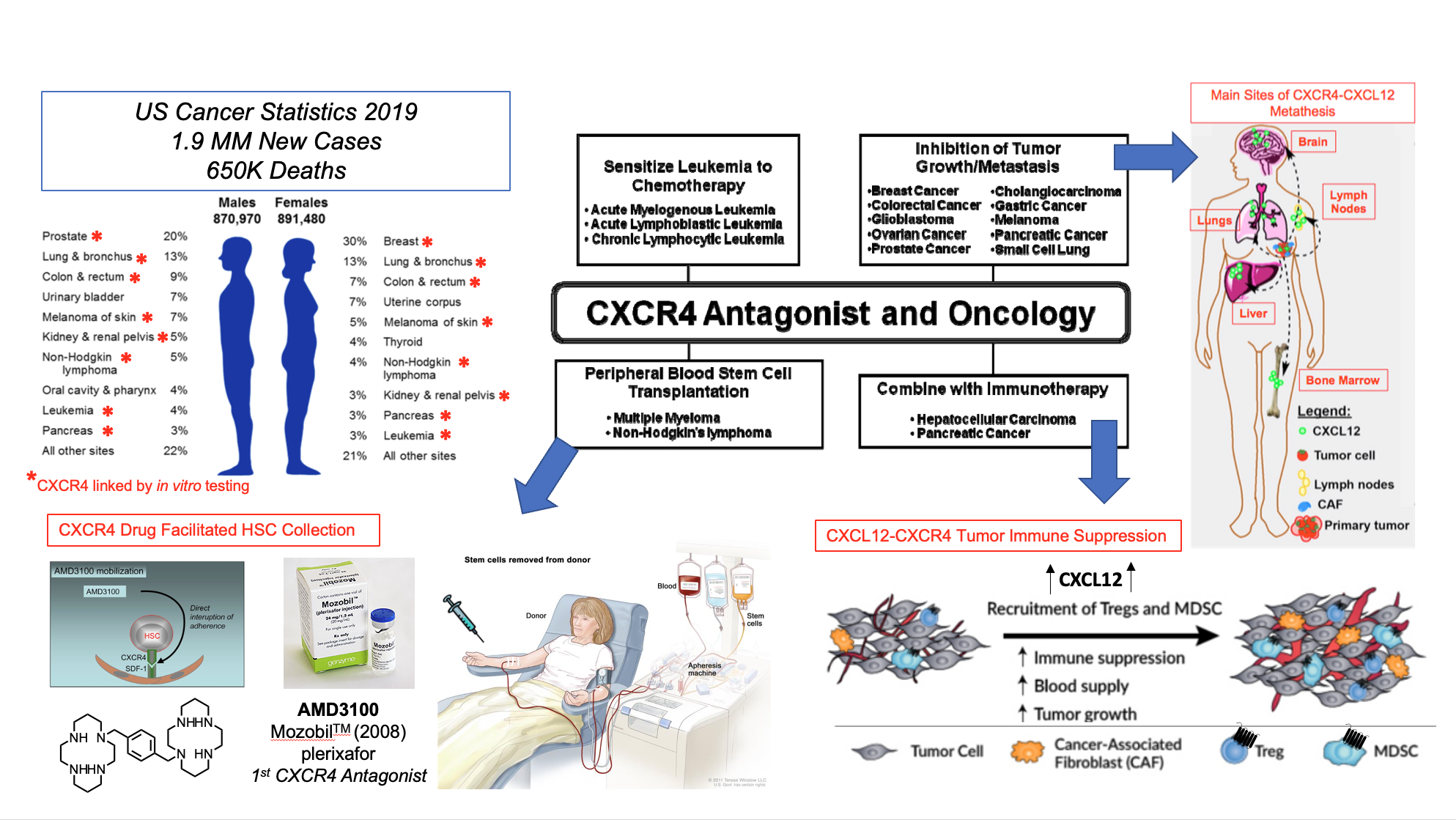
The over expression of CXCR4 on > 48 cancer cell types confers:
- Pro-survival gene transcription in the primary tumor microenvironment
- Pro-angiogenic and pro-vasculogenic cytokine expression promoting tumor progression and CXCR4+ cancer cell access to systemic circulation
- Immunosuppressive and chemo-resistant adhesion to and migration beneath tumor-associated stroma
CXCR4 and CXCL12 are independent indicators of poor clinical prognosis for patients suffering from various types of cancer.
Approaches to treating cancer that involve CXCR4 related mechanisms include stem cell transplantation for leukemias and myelomas, direct reduction of tumors by interference with oncogenic mechanisms and interference with cancer immune resistance.
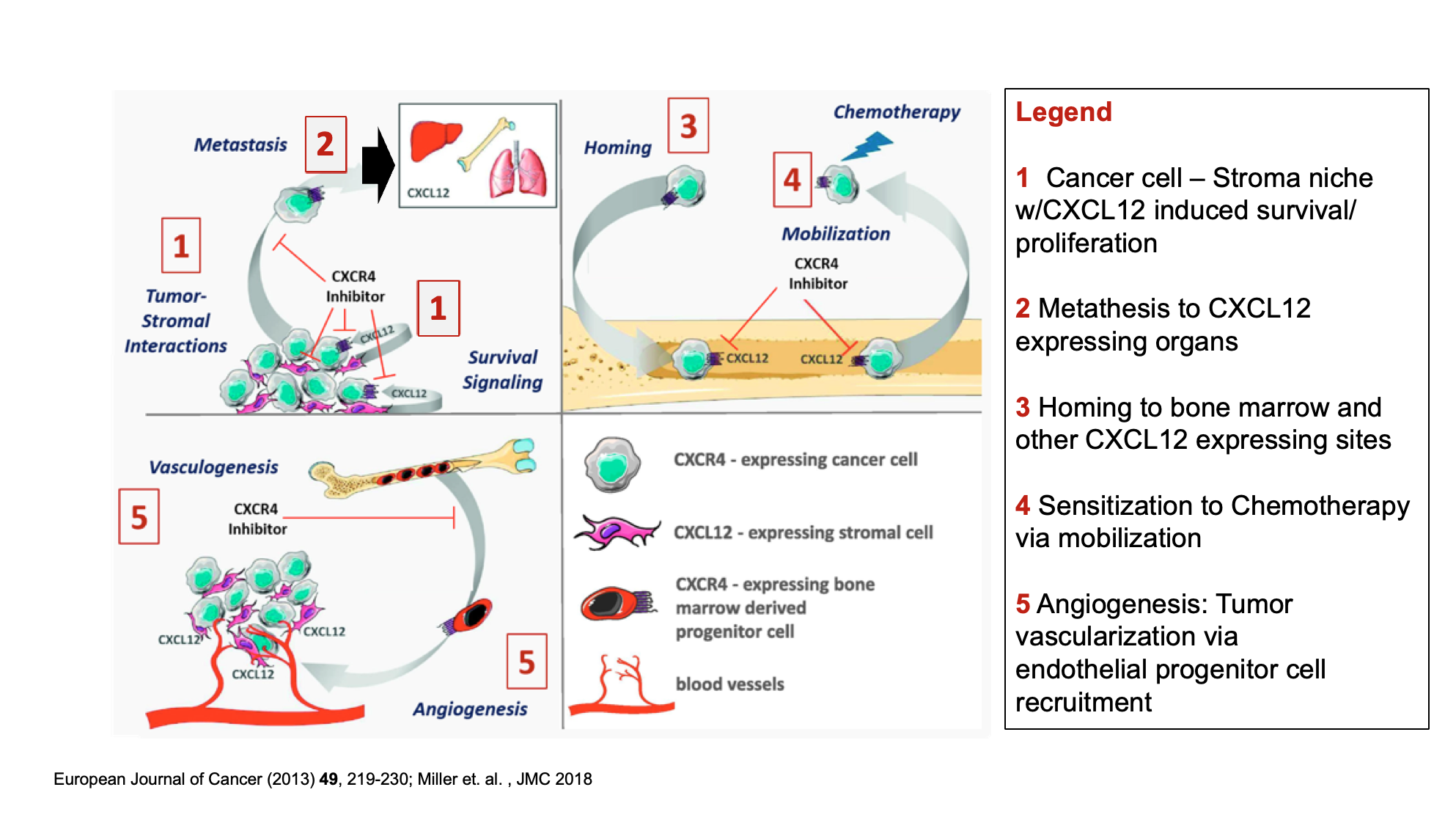
CXCR4 antagonists can inhibit cancer progression by:
- Disrupting tumor-associated stromal interactions that confer tumor cell survival and drug resistance
- Blocking the autocrine and paracrine growth and survival signals through activation of the CXCR4/CXCL12 axis
- Hindering tumor cell metastasis and homing to distant CXCL12-rich stromal niches
- Tumor cell mobilization from CXCL12-rich tissue to increase chemotherapeutic accessibility
- Obstructing CXCL12-mediated recruitment of pro-vasculogenic and pro-angiogenic CXCR4+ bone marrow-derived progenitor cells
- Sensitizing tumors to existing chemotherapy
These are summarized in the following diagram:
Another area of interest involves treating immune deficiencies. One of the most under treated and least acknowledged of these is the condition known as neutropenia, which is a deficiency in circulating neutrophils. Neutropenia patients suffer from various conditions including an increased risk of life threating bacterial and fungal infections. Neutropenia patients as a result are more likely to be hospitalized and given additional medications. CXCR4 plays an important role in both the maturation of new neutrophils via acceleration of the switch from CXCR4 to CXCR2 receptor populations and blockade of neutrophil recycling.
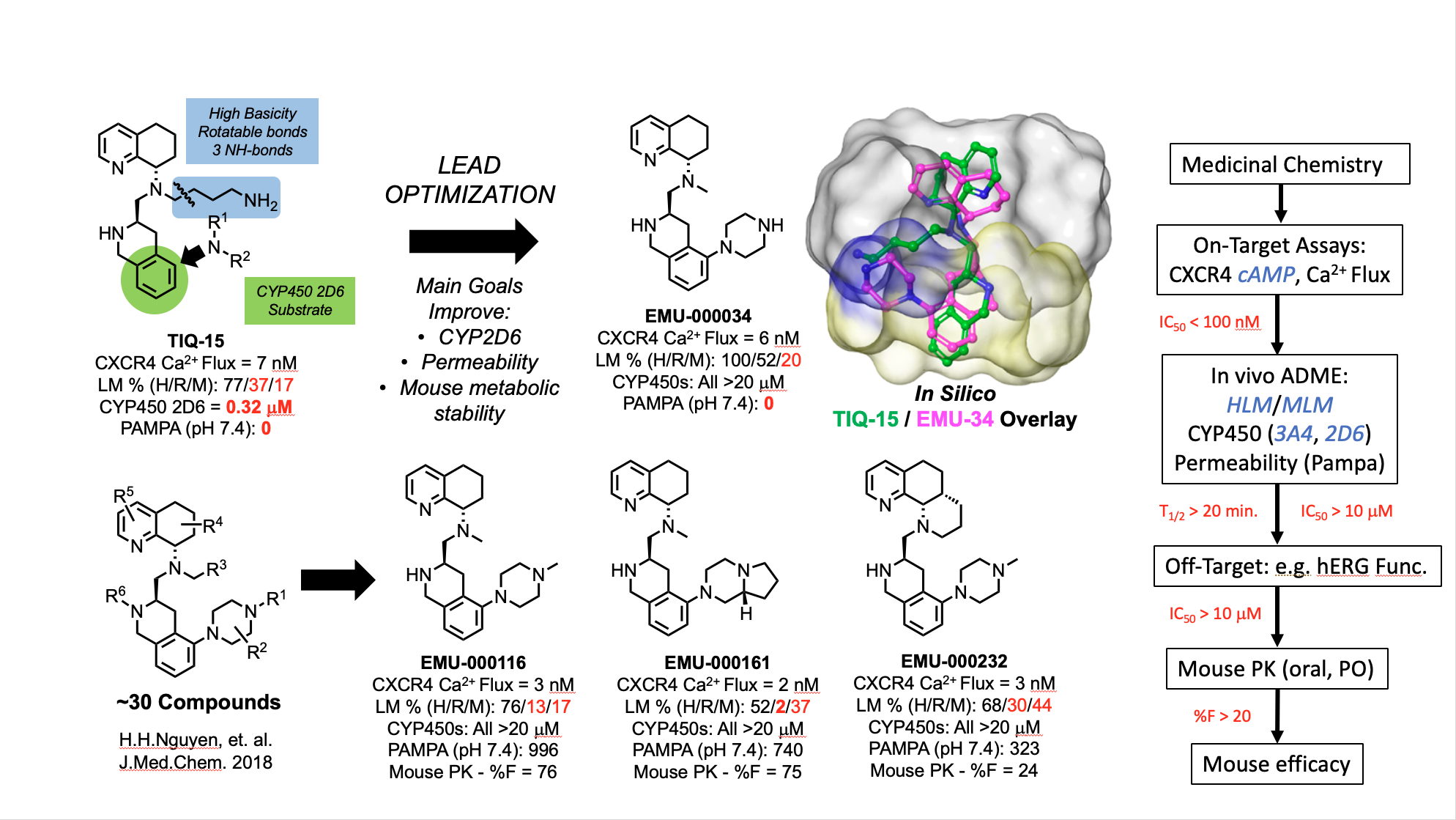
The evolution of our tetrahydroisoquinoline-based CXCR4 antagonists began with the discovery of the butyl amine compound TIQ-15. A focused lead optimization program led to the discovery of a second generation series of compounds with 2-piperazinyl substitutions on the TIQ ring. The design of these compounds was based on conformational analysis with a SAR focus (see overlay of TIQ-15 and EMU-34) and improvement of in vitro biochemical and biophysical properties (permeability, metabolic stability). A streamlined list of actions to evaluate both the in vitro an in vivo drug properties has resulted in identification of a clinical candidate (EMU-116) and several backup compounds (EMU-161 &232).
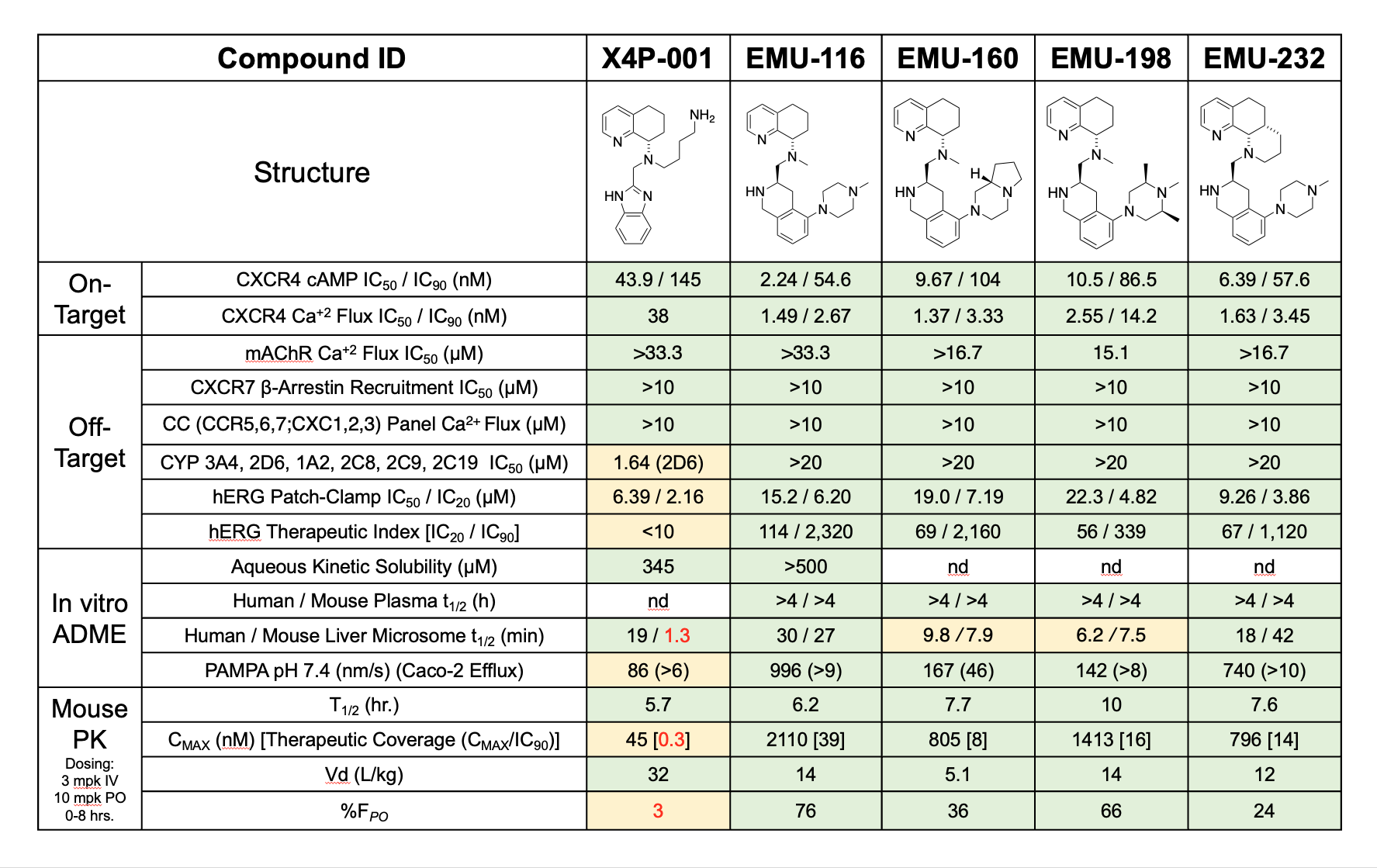
The in vitro activity profiles of the best lead compounds are shown below. Most notably, clinical candidate EMU-000116 demonstrates superiority over X4P-001, which is currently undergoing Phase 2 and Phase 3 clinical evaluation. As highlighted in the table, EMU-000116 outperforms X4P-001 in human liver microsomal stability assays, PAMPA permeability assays, and aqueous solubility assays. EMU-000116 also exhibits a much more attractive therapeutic index, as indicated by the ratio of cAMP IC90 values to hERG IC20 values.
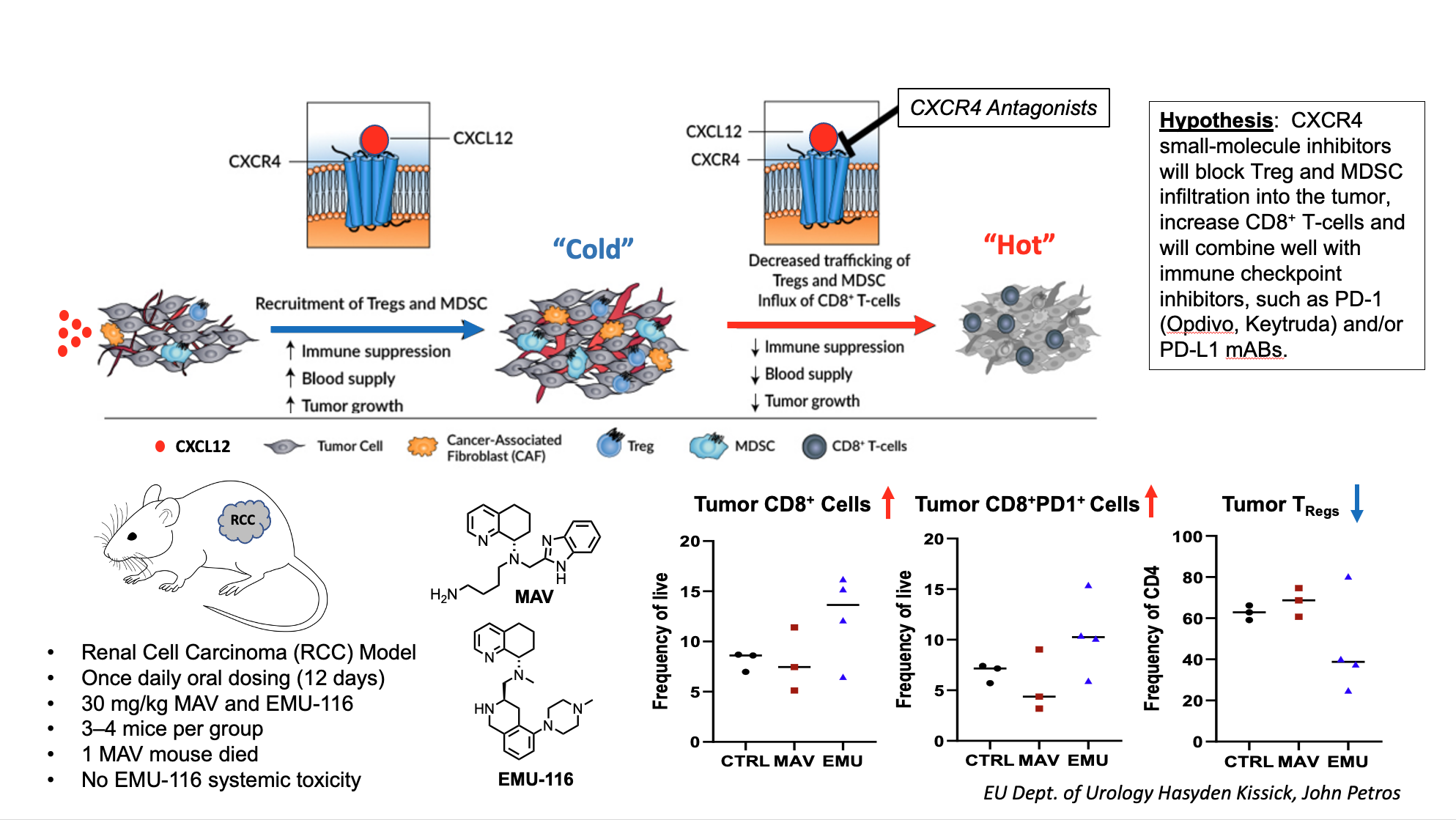
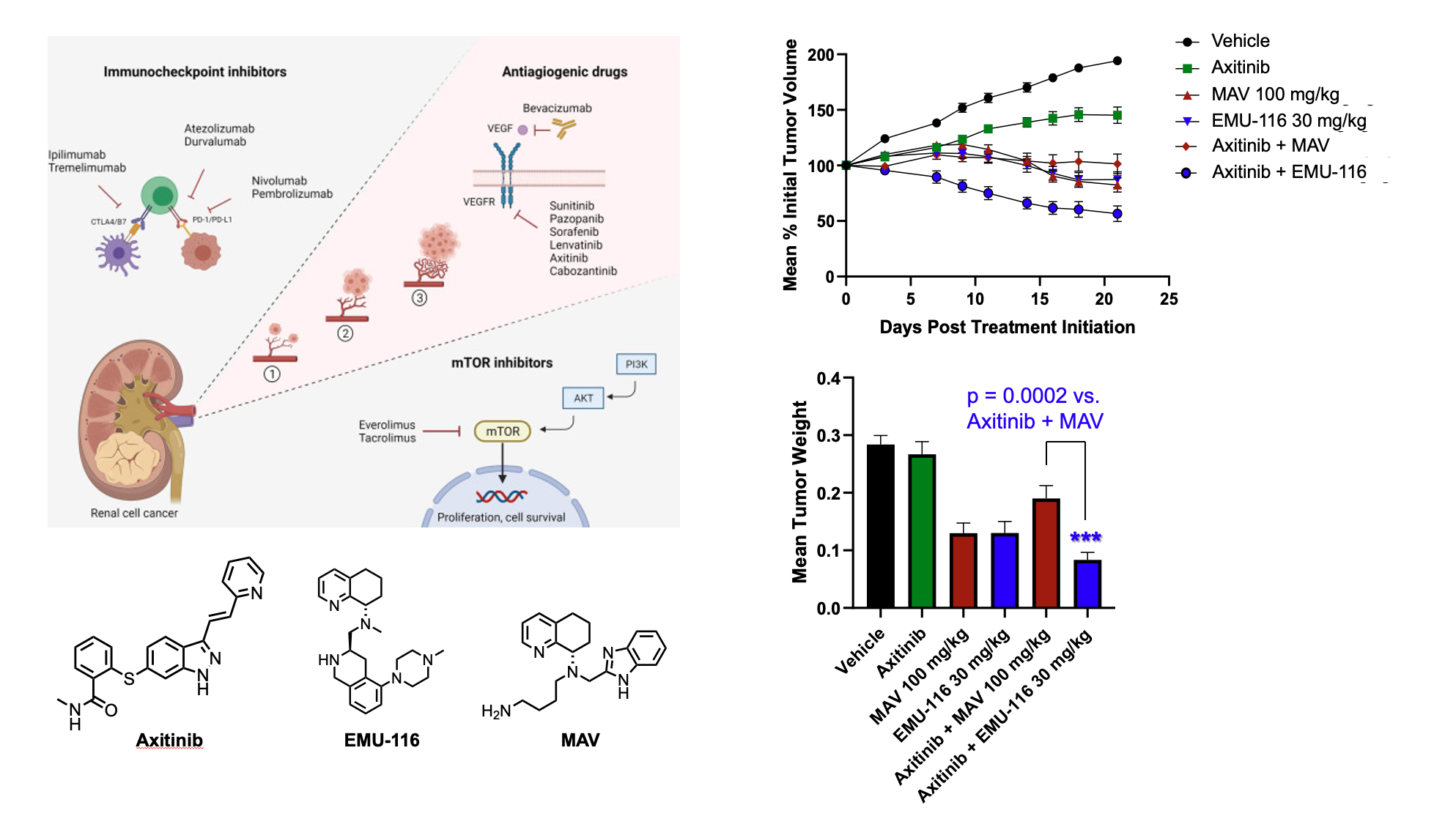
In a test of CXCR4 tumor-immune system engagement, a mouse renal cell carcinoma was used to evaluate the concept of turning cold tumors hot. In this example, tumors which are immune system deactivated (cold) from immune suppression result in tumor growth and increased blood supply mainly caused by the repopulations of T-cell subsets, mainly T-regulatory cells. CXCR4 antagonists will readjust tumor micro-environments by reducing immune suppression through enhancing CD8 T-cells and reducing Treg cells making them active (hot) for either immune or chemotherapy. Here a mouse with renal cell carcinoma was dosed with our lead compound EMU-116 and the current clinical agent Mavorixafor (MAV, X4P-001) and increase in CD8 T-cells was observed with concomitant reduction Tregs.
To examine whether these alterations in cell components would translate to improved efficacy, EMU-000116 was compared head-to-head to X4P-001 in a mouse xenograft model of renal cell carcinoma. In this case, a combination with a known anti-angiogenic RCC drug axitinib (3mg/kg fixed dose) was performed. Here, the 10 mg/kg dose of EMU-000116 was equally effective as 100 mg/kg X4P-001 at combating tumor growth. Furthermore, the 30 mg/kg EMU-000116 not only statistically significantly demonstrated superior anti-tumor efficacy relative to 100 mg/kg X4P-001, but tumor size was actually reduced under this therapeutic paradigm. Furthermore, tumor reductions were also observed by dosing the CXCR4 antagonists alone. Notably, none of the dosing regimens caused systemic toxicity, as indicated by the lack of changes in body weight over time.
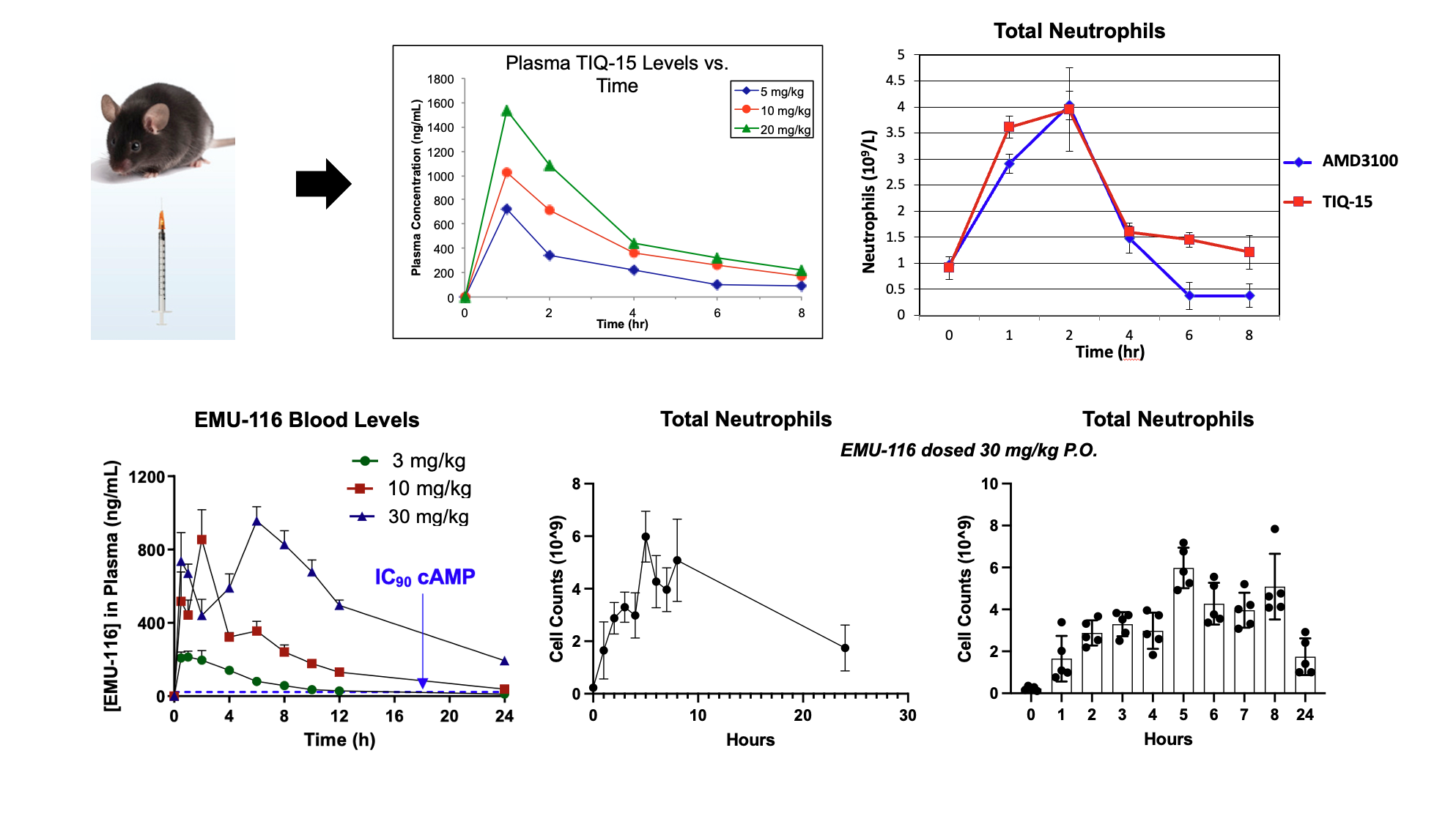
One other area of interest has been in the treatment of neutropenia. The distinct mechanisms involving CXCR4 in neutrophil regulation and populations has strong scientific correlation. To evaluate this effect, we simply dosed mice with our CXCR4 lead compounds (TIQ-15 & EMU-116) and then measured neutrophil counts in the blood via CBC analysis. Original lead compound TIQ-15 was dosed sub-cutaneous (S.C.; 5, 10 and 20 mg/kg) and compared to AMD3100, and EMU-116 was dosed orally (P.O.; 30 mg/kg). The results show that TIQ-15 and EMU-116 both mobilize and increase neutrophil cell counts in a dose-dependent manner. Both TIQ-15 and AMD3100 mobilize neutrophils in a comparable quantity when both are dosed S.C. while EMU-116 dosed orally provided an overall best response peaking at ~6 X 109 cells/L versus for both TIQ-15 and AMD3100 the quantity is ~4 X109 cells/L.